MECP2 duplication syndrome is a rare Xlinked neurodevelopmental disorder caused by an extra copy of theMECP2gene. It leads to earlyonset hypotonia, feeding difficulties, recurrent infections, and progressive neurological decline. In this article youll get straight answers about the symptoms, diagnosis, treatment options, life expectancy, and how it differs from Rett syndromeall written in a friendly, downtoearth style.
Stick with me and youll walk away with a clear picture of what families and clinicians face, plus practical tips and reliable resources you can use right away.
Quick Clinical Overview
What causes MECP2 duplication syndrome?
The condition stems from a duplication of a segment on the Xchromosome that includes theMECP2gene.MECP2produces a protein that helps regulate other genes in the brain; too much of it disrupts normal neuronal development. The duplication can vary in size, but most patients have a segment that also includes neighboring genes, which can influence severity.
How common is it?
Estimates suggest a prevalence of roughly 1 in 150,000250,000 live births, making it how rare is MECP2 duplication syndrome? a question many families wonder about. Although rare, awareness is growing thanks to better genetic testing and international registries.
Who is most affected?
Because the gene sits on the X chromosome, males are predominantly affected. Females can be carriers and, in rare cases, develop symptoms when Xinactivation is skewed. A few male patients survive into adulthood, often thanks to intensive multidisciplinary care.
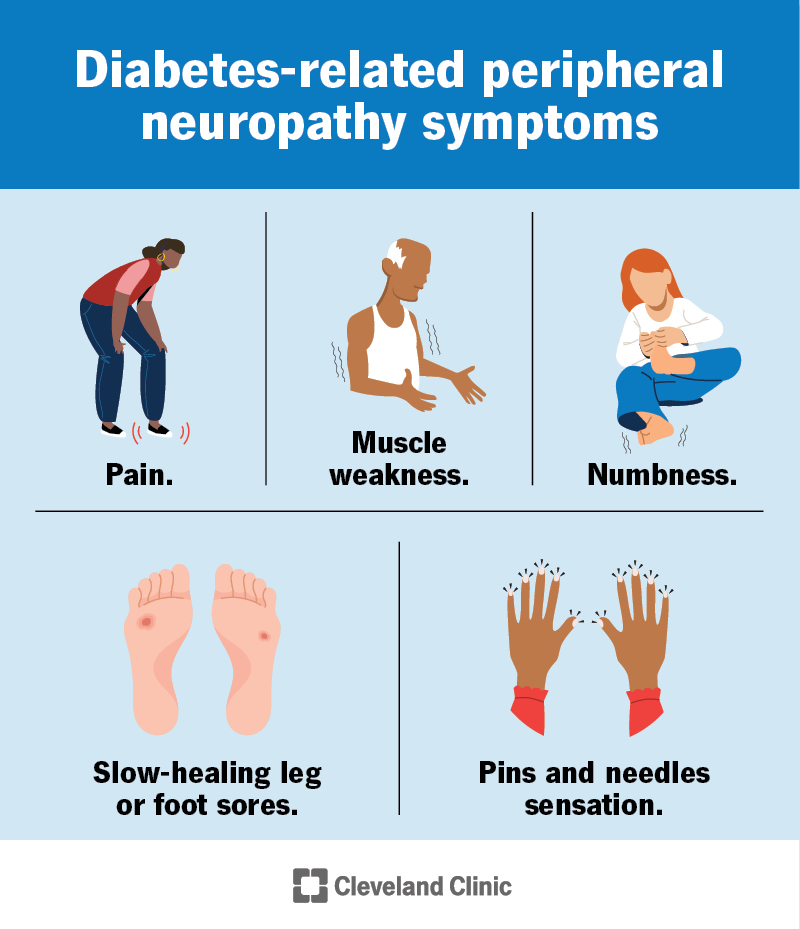
Key Symptoms Overview
What are the hallmark signs?
The most common early signs include:
- Severe hypotonia (floppy baby syndrome)
- Feeding difficulties, sometimes requiring a Gtube
- Recurrent respiratory infections and chronic lung issues
- Developmental delay and intellectual disability
- Seizures, which may begin in early childhood
These symptoms can appear as early as the first few months of life, so clinicians often suspect the disorder when routine checkups reveal a combination of weakness, poor weight gain, and persistent infections.
What about agespecific symptom patterns?
During infancy, hypotonia and feeding problems dominate. By preschool age, motor milestones lag behind peers, speech may be limited, and behavioral challenges (including autismlike traits) become apparent. In adolescence, some individuals develop scoliosis, constipation, and worsening seizure control.
Are there any rare or extra features?
Yessome patients exhibit immunodeficiency, leading to unusually frequent or severe infections. Others may develop constipation, scoliosis, or even cardiac anomalies. These extra signs reinforce the need for a comprehensive evaluation.
Quick symptom checklist
| Symptom | Typical Onset | Notes |
|---|---|---|
| Hypotonia | Birthto3months | Often the first clue |
| Feeding difficulty | First few months | May need Gtube |
| Respiratory infections | Infancy onward | Related to immunodeficiency |
| Developmental delay | 12years | Variable severity |
| Seizures | Variable, often <3years | Requires EEG monitoring |
| Immunodeficiency | Early childhood | Consult immunology |
Diagnosis and Testing
When should a clinician suspect MECP2 duplication?
If a male infant presents with floppybaby signs, feeding problems, and a history of recurrent infections, a geneticist should be consulted early. The same suspicion applies when a child shows unexplained developmental regression paired with seizures.
Which genetic tests confirm the diagnosis?
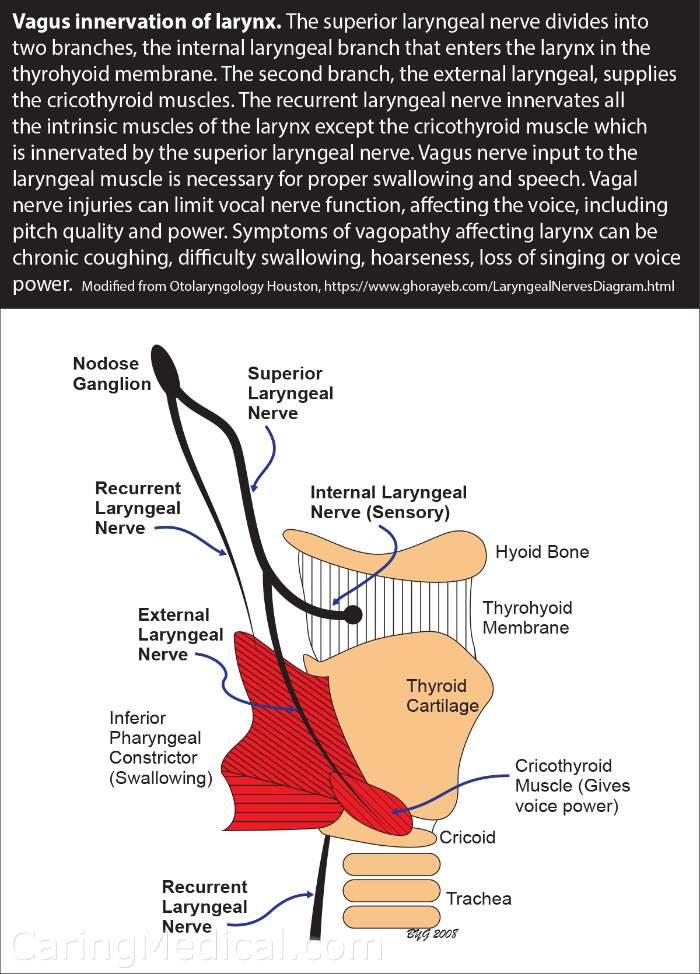
Chromosomal microarray analysis (CMA) is the frontline test; it can detect copynumber variations, including the MECP2 duplication. For finer resolution, multiplex ligationdependent probe amplification (MLPA) or nextgeneration sequencing (NGS) panels targeting neurodevelopmental disorders are also useful. The condition is coded in the ICD10 system asQ93.4(MECP2 duplication syndrome), which helps with insurance billing and epidemiologic tracking.
How is it differentiated from Rett syndrome?
Rett syndrome, caused by lossoffunction mutations in the same gene, presents mainly in females and usually features a period of normal development followed by regression. In contrast, MECP2 duplication syndrome often begins with hypotonia and never shows the classic "handwringing" behavior of Rett. The table below highlights the key differences.
| Feature | MECP2 Duplication | Rett Syndrome |
|---|---|---|
| Genetic mechanism | Extra copy (gainoffunction) | Lossoffunction mutation |
| Typical gender | Mostly males | Mostly females |
| Onset | Birthtoearly infancy | 618months after normal development |
| Key symptoms | Hypotonia, feeding issues, immunodeficiency | Handwringing, loss of speech, gait abnormalities |
| Life expectancy | Late teensearly 20s (improved with care) | Variable, often into adulthood |
Treatment and Management
Is there a cure?
Unfortunately, there is no cure for MECP2 duplication syndrome at the moment. The focus is on supportive care that mitigates complications and maximizes quality of life.
What does standard care look like?
Multidisciplinary management is the gold standard. Typical components include:
- Physical & occupational therapy to improve muscle tone, posture, and functional skills.
- Speech and feeding therapy essential when oral intake is unsafe; many families transition to Gtube feeding.
- Seizure control tailored antiepileptic regimens, often requiring regular EEG monitoring.
- Immunology support prophylactic antibiotics or immunoglobulin replacement for those with immunodeficiency.
- Respiratory care aggressive treatment of infections, sometimes using home ventilation.
Early intervention services can make a noticeable difference, especially for motor milestones and communication.
Any promising therapies on the horizon?
Researchers are exploring genesilencing approaches, such as antisense oligonucleotides, to reduce the extra MECP2 copy. A 2024 review in highlighted earlyphase trials showing modest improvement in mouse models. While human data are still emerging, families can keep an eye on clinical trial registries for upcoming studies. For families navigating treatment costs and coverage questions related to new therapies, resources about Exondys 51 insurance can provide useful examples of how specialty drug coverage discussions are structured.
How to manage immunodeficiency?
Patients with recurrent infections often benefit from monthly immunoglobulin (IVIG) infusions and vigilant vaccination schedules. Collaboration with an immunologist helps tailor prophylaxis and monitor antibody levels.
Life Expectancy Outlook
What is the typical life expectancy?
Historically, many individuals did not survive past early adolescence due to severe respiratory complications. Recent advances in multidisciplinary care, vigilant infection control, and seizure management have pushed the median survival into the late teens or early twenties for many patients. Of course, individual outcomes vary based on the severity of symptoms, access to specialized care, and family support.
How can families improve longterm outlook?
Key factors include early diagnosis, proactive respiratory therapy, consistent seizure control, and routine immunology followup. Community support groups also provide emotional bolstering, which can indirectly improve adherence to care plans.
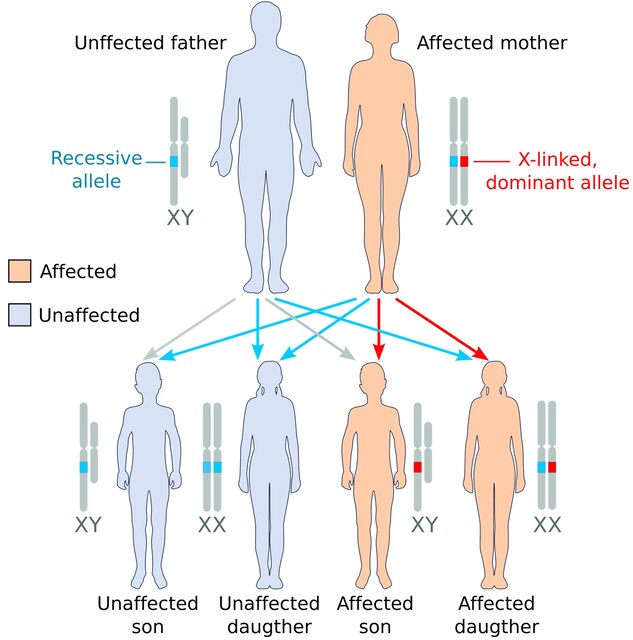
Impact on Females
Why are females less commonly affected?
Females have two X chromosomes, so one normal copy often compensates for the duplicated one. However, when Xinactivation (the process that silences one X chromosome in each cell) is skewed toward the normal X, the duplicated MECP2 becomes more active, leading to symptoms.
How do symptoms differ in females?
When females do manifest the syndrome, they tend to have milder hypotonia and lateronset developmental delays. Some may experience learning difficulties and subtle motor issues, but severe respiratory problems are less common. Genetic counseling is crucial for carrier females planning families, as the duplication can be transmitted to sons who will be more severely affected.
Duplication vs Rett Comparison
What are the core genetic contrasts?
Both disorders revolve around the same gene, yet the direction of the genetic change flips the clinical picture. Too much MECP2 (duplication) pushes the nervous system into a hyperregulated state, while too little (Rett) leads to underregulation. This mirrors the classic Goldilocks principle: the right amount is just right.
What practical tips help differentiate them in the clinic?
Ask these quick questions:
- Is the patient male or female?
- Did symptoms appear at birth (duplication) or after a period of normal development (Rett)?
- Is there a history of immunodeficiency and recurrent infections?
Answers guide the genetic testing strategy and counseling approach.
Coding and Helpful Resources
What is the ICD10 code?
The official ICD10 classification for MECP2 duplication syndrome is Q93.4. Using the correct code ensures proper documentation, insurance reimbursement, and facilitates research data collection.
Where can families find reliable information?
Trusted sources include:
- detailed medical overview and management guidelines.
- MECP2 Duplication Foundation patient stories, support groups, and uptodate research alerts.
- Childrens Hospital of Philadelphia (CHOP) specialized care pathways for rare genetic neurodevelopmental disorders.
How to connect with the community?
Online forums, Facebook groups, and annual family meetings organized by the MECP2 Duplication Foundation provide a safe space to exchange experiences, ask questions, and get emotional support. Many families also join research registries, which help scientists gather data for future therapies.
RealWorld Perspectives (Experience)
Case study: A familys journey
When Jamies parents first noticed his constant floppiness and difficulty feeding, they were overwhelmed. After several pediatric visits, a geneticist ordered a CMA that revealed the MECP2 duplication. Within weeks, Jamie began a coordinated care plan: a speech therapist helped with oral feeds, a neurologist started seizure prophylaxis, and an immunologist began monthly IVIG. Two years later, Jamie is thriving in a mainstream classroom, using a communication device, and his family reports a far better quality of life than they imagined possible.
Clinician insight
Dr. Lena Morales, a pediatric neurologist at a leading childrens hospital, emphasizes early multidisciplinary involvement is the single most impactful factor for these kids. She notes that families who receive clear, empathetic explanations early on tend to stay more engaged and experience fewer emergency hospitalizations.
Conclusion
MECP2 duplication syndrome is a rare but serious Xlinked condition that brings unique challengesfrom early hypotonia and feeding struggles to immunodeficiency and seizures. While there is no cure yet, a proactive, multidisciplinary approachgrounded in accurate diagnosis, vigilant infection control, and personalized therapycan extend life expectancy and dramatically improve daytoday quality. By staying informed, connecting with trusted resources, and leaning on supportive communities, families can navigate the journey with confidence and hope. If you or someone you love is dealing with this diagnosis, consider reaching out to the MECP2 Duplication Foundation for uptodate research news and peer supportyoure not alone in this.